

Minéralogie
La minéralogie a pour but l'étude des minéraux, éléments ou composés naturels. Si la notion d'espèce minérale a longtemps varié, depuis quelques décennies les minéralogistes en donnent la définition suivante : un minéral est un solide naturel, homogène, possédant une composition chimique définie et une structure atomique ordonnée. Il convient de développer cette définition.
Solide : à l'exception du mercure natif, aucun liquide n'est considéré comme un minéral.
Naturel : il est possible de reproduire artificiellement des minéraux : historiquement commencée avec les pierres précieuses artificielles, la synthèse des cristaux (quartz, diamant) est devenue une industrie importante ; mais ces produits résultant de l'activité humaine ne sont absolument pas des minéraux au sens vrai du terme.
Homogène : un minéral est composé d'une seule phase solide et aucune méthode physique de séparation ne peut permettre d'en isoler des composés plus simples ; de nombreuses espèces minérales se sont révélées être des mélanges quand on a pu disposer de méthodes précises d'examen.
Composition chimique définie : les exceptions à cette règle proviennent de l'existence de solutions solides permettant des variations de composition entre deux ou plusieurs termes extrêmes et de possibilités de remplacement d'un élément par un autre.
Structure atomique ordonnée : un minéral est un solide cristallisé, constitué par un arrangement périodique d'atomes d'éléments donnés ; par exception, on considère comme minéraux des verres et des gels solidifiés (opale, chrysocolle, allophanes), et aussi les espèces « métamicte », dont la structure a été plus ou moins complètement détruite par les rayonnements du thorium ou de l'uranium.
Les minéraux, sauf de rares exceptions (oxalates, mellates, citrates...), sont des composés inorganiques. Les houilles, les hydrocarbures solides sont des roches, des mélanges qui peuvent être séparés physiquement en constituants plus simples.
La minéralogie doit donc recenser, reconnaître, définir les propriétés et les conditions d'apparition et d'évolution des espèces minérales. On distingue aujourd'hui une minéralogie « industrielle », qui doit promouvoir l'utilisation des minéraux, contribuer aux techniques de la valorisation des minerais, étudier les méthodes de synthèse des espèces trop rares dans la nature pour la demande industrielle.
Les roches, les minerais, les sols étant surtout d'origine minérale, le rôle et l'importance de la minéralogie dans les sciences de la Terre sont évidents. Mais elle a aussi apporté son concours à d'autres disciplines : chimie, métallurgie, physique du solide. Il suffit de rappeler que les techniques radio cristallographiques viennent directement des recherches minéralogiques : qu'en serait-il de la physique moderne sans cet apport ?
La minéralogie se situe donc au carrefour des sciences d'observation et des sciences exactes et. à ce titre, elle est une discipline étroitement liée au développement de la civilisation actuelle.
Les différents domaines de la minéralogie découlent de la définition même de l'espèce minérale :
de la géométrie du réseau et de la nature des atomes le constituant dérivent la cristallographie, la cristallochimie, la physicochimie du solide naturel, la minéralogie descriptive et déterminative, la minéralogie industrielle.
L'étude des conditions de formation et d'évolution des minéraux constitue la minéralogie génétique et une partie importante de la géochimie.
Après avoir esquissé les grands traits de l'évolution de la minéralogie, on traitera de l'important domaine de la cristallochimie pour ce qui concerne l'étude de la géométrie et des propriétés du milieu cristallin) ; puis seront exposés les caractères permettant d'identifier les espèces minérales ; enfin, on abordera les principes de leur classification, en accordant un développement particulier aux classes qui ne font pas l'objet d'articles spéciaux.
1. Historique
Ce sont des minéraux - éléments natifs comme le cuivre, l'or - qui ont été les premiers métaux utilisés par l'homme. En particulier, le cuivre a ouvert l'ère des métaux avec l'âge du cuivre ou Chalcolithique (vers 6000 av. J.-C).
Aristote divisa en deux classes le monde minéral : les métaux et les « fossiles » (roches et minéraux non métalliques). On trouve ensuite les travaux des naturalistes arabes, et en particulier d'Avicenne dont la classification des minéraux en quatre groupes - pierres et gemmes, minerais, combustibles, sels - se retrouvera jusqu'au XIXe siècle.
G. Agricola (1494-1555 ) peut être considéré comme le père de la minéralogie moderne ; il sut en effet tourner résolument le dos à l'alchimie et observer les minéraux dans leurs gisements, posant ainsi les fondements de la métallogénie et de la géologie minière. Il utilise dans ses œuvres (surtout De natura fossilium, 1546, et De remetallica, 1556) les propriétés immédiates des minéraux (couleur, éclat, dureté, densité, goût, odeur...) pour les décrire.
Au XVIIe siècle, N. Stenon (1669) montre la constance des angles des faces cristallines pour certaines espèces définies. De ce siècle date aussi l'optique cristalline avec E. Bartholin (double réfraction) et les célèbres travaux de C. Huygens.
La fin du XVème siècle vit l'essor de la cristallographie.
En 1783, Rome de l'Isle montre que les angles dièdres que font entre elles les faces d'un cristal sont constants pour tous les cristaux d'une même espèce. L'abbé R. J. Haûy, le père de la cristallographie, publia en 1784 son Essai d'une théorie sur la structure des cristaux, énonça la loi des troncatures rationnelles, découvrit les règles de symétrie, distingua les sept systèmes cristallins et la dérivation des formes secondaires.
Au début du XIXe siècle, l'aspect chimique de la minéralogie se développa considérablement en participant à l'essor de la chimie minérale. J. J. Berzelius (1779-1848) et E. Mitscherlich (1794-1863) étudièrent la chimie des espèces minérales, ce qui permit à ce dernier de découvrir l'isomorphie et la polymorphie.
Malheureusement, sous l'influence dogmatique de Berzelius, la composition chimique devint, pour de trop longues années, la seule base de la notion d'espèce minérale, malgré les efforts d'Haùy, qui, avec une vision exceptionnelle de la réalité, chercha à lier structure et composition. C'est de cette époque « chimique » que datent de nombreuses espèces ou variétés sans existence réelle, dues à des analyses imparfaites ou effectuées sur des mélanges.
R. J. Haiiy avait montré l'existence d'un réseau cristallin triplement périodique, J. F. C. Hessel et A. Bravais étudièrent les 32 groupes de symétrie, bientôt complétés par les travaux de A. M. Schônfliess et V. S. Fedorov sur les 230 groupes spatiaux.
En optique cristalline, il faut citer E. Mallard, F. Becke, V. S. Fedorov. H. C. Sorby qui, en introduisant la méthode des lames minces et des sections polies, permirent le développement de la pétrographie (K. H. F. Rosenbush, A. Michel-Lévy, A. Lacroix) et de la microscopie des minerais (H. Le Chatelier, P. Ramdhor).
Au point de vue chimique, il est difficile de dissocier les progrès de la minéralogie du développement de la chimie minérale. A. Daubrée (1814-1896) et son école de géologie expérimentale sont à la base des études actuelles sur les synthèses et les conditions d'équilibre des minéraux.
Si C. F. Schônbein employa, en 1838, le terme de géochimie, cette discipline devait attendre le début du XXe siècle pour trouver une réelle individualité, grâce aux travaux de F. N. Clarke, V. M. Goldschmidt, V. I. Vernadsky et A. Fersman.
Mais l'événement le plus marquant fut la réalisation, par M. von Laue en 1912, de la diffraction d'un faisceau de rayons X par un cristal de blende ; avec la structure périodique de la matière, atomes et molécules devenaient une réalité. Ces travaux et ceux de W. H. et W. L. Bragg, P. Debye, posaient les bases de la cristallochimie et permettaient enfin une définition rigoureuse de l'espèce minérale.
2. Cristallochimie
La cristallochimie peut être définie comme l'étude des relations existant entre la composition chimique d'un solide, l'arrangement géométrique et les forces de liaison entre les atomes constituant le réseau. Ces relations déterminent les propriétés physiques et chimiques. La cristallochimie permet donc de définir les propriétés d'une substance en fonction de sa structure.
Forces de liaison et stéréochimie
On peut définir pour les atomes des volumes d'encombrement, ce qui permet de les représenter par des sphères dont le rayon, de l'ordre de l'angstrôm (0,10 nm), peut être mesuré avec une grande précision par les méthodes radio cristallographiques. Ces rayons atomiques varient avec l'élément, son état d'ionisation, la nature de ses liaisons avec les atomes voisins.
Liaisons atomiques
Dans un cristal, les liaisons et donc les forces par lesquelles s'attirent les atomes peuvent être de différente nature.
Les liaisons métalliques s'exercent entre atomes de métaux électropositifs. Les métaux natifs, de nombreux sulfures et sulfosels possèdent des structures basées sur ces liaisons.
Ils peuvent alors être considérés comme formés par un assemblage d'ions positifs, les électrons libérés étant dispersés et se déplaçant facilement au sein de cet assemblage ; pouvant interférer avec les rayons lumineux, ils rendent les métaux opaques et leur donnent leur éclat ; leur mobilité explique les bonnes conductivités thermiques et électriques.
La faiblesse des liaisons métalliques et leur facile reconstitution explique la fréquente malléabilité de ces espèces.
Les liaisons covalentes ou homopolaires existent entre les atomes non métalliques de même électronégativité, les atomes voisins mettant en commun leurs électrons de valence en complétant leur couche électronique périphérique.
Dans le diamant, par exemple, chaque atome de carbone est entouré par quatre autres atomes de carbone, situés au sommet d'un tétraèdre régulier circonscrit, chacun mettant en commun un électron avec l'atome central, dont la couche électronique de valence se trouve ainsi saturée.
Les liaisons ioniques ou polaires, de nature électrostatique, existent entre ions de charges contraires et obéissent à la loi de Coulomb. Dans ces liaisons, il se produit un transfert d'électron de l'élément électropositif vers l'élément électronégatif, chaque atome acquérant la configuration électronique d'un gaz rare. À l'exception des éléments et sulfures, les liaisons ioniques sont fréquentes dans le règne minéral.
Les liaisons de Van der Waals seraient dues à la résonance des états vibratoires des atomes ; elles existent dans toutes les structures mais leur intensité est très faible par rapport aux autres liaisons.
Dans les rares minéraux à réseaux moléculaires (soufre, réalgar AsS, calomel Hg2Cl2, sénarmontite Sb2O3), les liaisons intramoléculaires, covalentes, sont fortes, alors que les molécules sont faiblement liées entre elles par des forces de Van der Waals.
Des liaisons de types différents coexistent souvent dans les minéraux ; dans ce cas, les propriétés physiques comme la dureté, le point de fusion, la résistance mécanique sont fonction des liaisons les plus faibles, qui cèdent en premier.
Dans certaines structures ioniques, il y a apparition d'ensembles individualisés : radicaux simples, tels que (CO3)2-, (SO4)2-, (PO4)3-, ou polymérisés (structures des silicates, des borates).
Nombre de coordination
On appelle nombre de coordination ou, plus simplement, coordinence d'un atome, le nombre total d'atomes, de radicaux ou de molécules unis à cet atome central.
Dans un cristal ionique, les ions de petite taille, cations en général, s'entourent du nombre maximum de gros ions, anions en général, et ce nombre est fonction des tailles relatives de ces ions ; de plus, comme il est nécessaire que l'assemblage soit électriquement stable, donc neutre, la somme des charges électriques s'équilibre exactement.
Quand le terme « coordinence » est utilisé sans être précisé, c'est que l'on se réfère à la coordinence par rapport à l'oxygène, élément de loin prédominant dans la croûte terrestre (46,6 % en poids et 92 % en volume) et dont l'arrangement détermine une grande partie des structures minérales.
La coordinence varie en fonction du rapport entre les rayons des ions considérés (tabl. 1).
Un assemblage compact (fig. 1) peut être soit hexagonal compact, soit cubique compact (cubique à faces centrées).
fig. 1 - Assemblages compacts.
tabl. 1 - Coordinences et structures correspondant aux différents rapports entre les rayons des ions en présence.
|
Rcation/Ranion |
coordinence |
structure |
|
0,15 à 0,22 0,22 à 0,41 0.41 à 0,73 0,73 à 1 1 |
3 4 6 8 12 |
sommets d'un triangle équilatéral sommets d'un tétraèdre sommets d'un octaèdre sommets d'un cube assemblage compact |
Dans les structures ioniques, les coordinences 5, 7, 9, 10 et 11 sont rares. La plupart des cations ont une seule coordinence par rapport à l'oxygène ; d'autres comme Al, Ca, Na, Zn, etc. en ont deux, car le rapport de leur rayon au rayon de l'oxygène est proche de la limite séparant deux domaines de coordinence.
Pour l'aluminium, ce rapport (0,43) est proche de la limite entre les coordinences 4 et 6. Cette double coordinence conditionne la structure et l'existence de très nombreux silicates ; en effet, la coordinence dépendra en partie de la température et de la pression à laquelle a lieu la cristallisation (une élévation de température et une diminution de pression favorisent une faible coordinence).
En général, les coordinences ne sont pas exprimées dans les formules chimiques des minéraux, sauf pour différencier certains polymorphes. Par exemple, andalousite, disthène et sillimanite ont pour formule Al2SiO5, mais les atomes d'aluminium ont des coordinences différentes précisées en exposants : andalousite, A1VIA1V [O | SiO4] ; disthène, Alv12 [O | SiO4] ; sillimanite, A1V1 AlIV [O | SiO4].
Valence électrostatique
Les composés ioniques devant être électriquement neutres, si la charge d'un cation est Z et le nombre d'anions l'entourant n, la force de liaison ionique entre le cation et un de ces anions est Z/n : c'est la valence électrostatique de la liaison. Les structures dans lesquelles toutes les liaisons sont de force égale sont appelées isodesmiques.
Dans les cristaux anisoclesmiques, les anions auront des liaisons plus fortes avec le cation central qu'avec les autres atomes, doù l'apparition d'un radical : carbonates, sulfates, etc. Enfin dans les structures mésodermiques - cas des silicates -, la valence électrostatique égale la moitié de la valence chimique de l'anion.
Il faut bien distinguer valence chimique et valence électrostatique : le nombre des anions entourant un cation est déterminé par la valence électrostatique, dépendant de la coordinence, et non par la valence chimique. Cela explique que certains composés ne puissent exister dans la nature bien qu'ils aient des formules théoriques compatibles avec les règles de valence chimique.
Énergie réticulaire
L'énergie réticulaire d'un cristal ionique est l'énergie nécessaire pour disperser en ions séparés une masse de ce cristal correspondant à une mole. Cette énergie sera d'autant plus grande que la charge des ions est plus forte, que les ions sont plus petits et que l'assemblage est plus compact (cf. cristaux - Cristallochimie minérale).
D'une façon générale, l'ordre de cristallisation des minéraux dans une roche suit plus ou moins régulièrement la décroissance de l'énergie réticulaire de leurs réseaux. De plus, à une diminution de l'énergie réticulaire correspond une diminution de la dureté, une augmentation de la solubilité, un accroissement des distances interréticulaires.
Formule chimique des minéraux
Pour définir une espèce minérale, il est nécessaire d'en connaître la composition chimique. Pour cela, après une analyse qualitative complète, on effectuera, par diverses méthodes physicochimiques, une analyse quantitative qui donnera les proportions relatives des éléments constituants et permettra ensuite d'en tirer une formule qui indique les éléments présents et en quelles proportions ils se trouvent.
Les analyses quantitatives sont souvent fort malaisées, la plupart devant être réalisées sur des minéraux difficiles à trier et de composition parfois très complexe. Ce n'est que depuis relativement peu de temps que la formule de nombreux silicates est bien connue, surtout grâce aux progrès réalisés dans la connaissance de leur structure.
Expression et interprétation des analyses
On exprime habituellement les analyses en pourcentage en poids d'éléments, d'anhydrides ou d'oxydes pour les minéraux oxygénés ; en effet, l'oxygène n'est pratiquement jamais déterminé directement.
Lorsqu'un minéral oxydé contient des éléments non métalliques autres que l'oxygène (halogènes, soufre, etc.), la somme des pourcentages en poids qui doit être voisine de 100 (99,5 à 100,5 pour une macroanalyse correcte) pourra nettement dépasser ce chiffre ; en effet, de l'oxygène a été indûment compté en quantité équivalente à celle nécessaire pour saturer les atomes métalliques combinés au soufre ou aux halogènes. Il faudra donc retrancher le poids de l'oxygène en excès.
On peut prendre quelques exemples de calculs permettant d'établir la formule d'un minéral à partir de son analyse, comme celle qui figure au tableau 2.
tabl. 2 - Calcul de la formule d'un minéral à partir de son analyse.
|
anhydrides et oxydes |
pourcentages en poids (1) |
poids moléculaires (2) |
rapports moléculaires (3) |
nombre de « molécules » (4)
|
|
CuO p2O5 H2O
|
68,58 24,72 6,76 |
63,54 141,96 18,02 |
0,863 0,174 0,375 |
4,96 1 2,16 |
|
total
|
100,06 |
|
|
|
Les rapports moléculaires sont obtenus en divisant le pourcentage en poids par le poids moléculaire des oxydes ou anhydrides.
Les rapports moléculaires sont entre eux comme les nombres de molécules d'oxydes contenus dans le minéral : pour 174 molécules de P2O5 on aura 863 molécules de CuO (en réalité, il n'existe pas de molécules d'oxydes ou d'anhydrides, mais c'est une façon conventionnelle d'exprimer l'oxygène non dosé).
En considérant la colonne (3) et en prenant comme égal à 1 le plus grand commun diviseur, soit 0,174, on obtient en (4) le nombre de « molécules » d'oxydes ou d'anhydrides constituant la formule, soit 4,96 CuO, P2O5, 2,16 H2O ; pour obtenir une formule stœchiométrique tenant compte de la loi de Dalton, on ramènera les exposants à des nombres entiers, soit : 5 CuO, P2O5, 2 H2O (pseudo malachite).
Mais, actuellement, les formules ne sont plus données sous forme dualistique mais structurale ; ces formules structurales indiquant à la fois les relations structurales des atomes entre eux et les remplacements possibles dans ces structures ; on aura donc Cu5(PO4)2, et les quatre valences positives restantes saturées par 4 hydroxyles, soit Cu5(PO4)2(OH)4.
L'eau pose souvent un problème dans les analyses ; elle peut exister sous forme adsorbée (humidité), ou sous forme d'eau de cristallisation, ou encore être formée à partir d'hydroxyles, d'ions hydrogène ou hydroxonium (H3O)+ existant dans la structure.
Il faut donc établir sous quelle forme elle se trouve par des méthodes annexes (analyse thermique, spectrographie infrarouge, etc.). Par convention, on désigne par H2O+ l'eau de constitution et par H2O l'eau d'adsorption, éliminée par chauffage à 110 °C et dont on ne tiendra pas compte dans les calculs analytiques et la formule.
Utilisation des formules structurales
Dans les minéraux, on l'a vu, la structure est pratiquement déterminée par l'arrangement des gros ions, anions en général, et pour les composés oxygénés par celui des ions oxygène ; si les méthodes radio-cristallographiques ont permis de déterminer dans quel groupe de structure connue se situe le minéral, par exemple pyroxène ou grenat, il est possible de calculer l'analyse sur la base du nombre d'atomes d'oxygène déterminant la maille et donc d'obtenir des formules structuralement plus exactes.
Utilisation des paramètres de la maille
On calcule le volume V de la maille en nm3 à partir des paramètres en nm (tabl. 3).
tabl. 3 - Volume de la maille en fonction de ses paramètres pour chaque système cristallin.
|
système |
volume de la maille |
|
cubique |
a3 |
|
quadratique |
a2c |
|
hexagonal |
a2c sin 60° |
|
rhomboédrique |
a3 (1 - cos α) (1 +2cos α) 1/2 |
|
orthorhombique |
abc |
|
monoclinique |
abc sin β |
|
triclinique |
abc (1 - cos2 α - cos2 β - cos2γ + 2 cos α cos (3 cos γ) 1/2 |
La densité mesurée étant d, le poids P du contenu de la maille élémentaire est égal à V x d (P en grammes = V.d.l024). Rappelons que la maille contient un nombre Z de « formules-unités » correspondant à la formule chimique usuelle. Le poids d'une « formule-unité » est égal au quotient de la masse atomique ou moléculaire M de la formule chimique usuelle par .V (0,602 3.1024). Le nombre Z se trouve donc égal à :

soit encore, si l'on exprime V en nm3 :
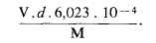
soit, si l'on exprime V en cm3 :

Détermination du nombre d'atomes dans la maille
Soit, pour une analyse de minéral, Z la somme des pourcentages en poids des éléments, oxydes, anhydrides du résultat analytique, en ne tenant pas compte de l'eau d'adsorption, Q le pourcentage par rapport à L d'un élément de masse atomique m, R = Q/m le rapport atomique de cet élément, d la densité mesurée, V le volume de la maille en nm3, et V.d.\0~24 le poids P en grammes du contenu de la maille. Le nombre Z d'atomes de l'élément considéré dans la maille est donné par :

Les phénomènes de remplacement dans les minéraux
Les résultats analytiques montrent que les espèces minérales ont rarement leur composition idéale. Les écarts constatés, souvent dus à des impuretés dans la prise d'essai, peuvent aussi résulter de substitutions dans les réseaux.
Les minéralogistes utilisent des formules indiquant les variations de la composition chimique, formules dans lesquelles tous les composants chimiques structuralement équivalents sont regroupés. Par exemple, l'almandin, le plus courant des grenats, a pour formule idéale :
Fe2+3 Al2[SiO4]3
mais on aura pour un almandin provenant d'une localité déterminée :
(Fe2+2,2 Mg0,4 Ca0,2 Mn0,2) (Al1,9 Fe1+0,3) [SiO4]3.
Deux espèces seront isostructurales si elles ont même formule chimique globale, même groupe de symétrie, mêmes coordinences des atomes les constituant.
Deux espèces seront isomorphes si elles sont isostructurales et si les rapports des rayons cationiques et anioniques des éléments les constituant sont assez proches pour que les deux espèces puissent plus ou moins complètement syncristalliser en donnant des cristaux de composition mixte.
Les cristaux mixtes résultant de la syncristallisation de composés isomorphes ou non sont appelés solutions solides.
Un terme de la série de solutions solides existant entre forstérite et fayalite (péridots) peut être considéré comme une solution homogène de Mg2[SiO4] dans Fe2[SiO4] (olivine) ; en réalité, il y a remplacement statistique dans le réseau d'ions Fe2+ par des ions Mg2+.
On appelle éléments diadochiques ceux qui peuvent se substituer les uns aux autres pour occuper de mêmes positions réticulaires dans un cristal. Dans la série forstérite-fayalite, Mg2+ et Fe2+ sont diadochiques. Les solutions solides peuvent former une série complète ou partielle entre les termes extrêmes.
On distingue trois types de solutions solides.
Dans les solutions solides de substitution, les atomes sont substitués diadochiquement ; pour cela il faut que l'atome substitué satisfasse à plusieurs conditions, et en particulier pour les cristaux ioniques :
- Les atomes ou ions peuvent se remplacer s'il n'y a pas plus de 15 % de différence entre leurs rayons ioniques pour une même coordinence. Par exemple, Cu2+ (r = 0,072 nm) et Zn2+ (r = 0,074 nm) pourront se remplacer facilement ; au contraire Ca2+ (r = 0,092 nm) et Mg2+ (r = 0,066 nm) n'auront que peu de possibilités de substitution bien que leurs propriétés chimiques présentent de nettes analogies.
- Normalement un ion ne peut être remplacé que par un ion de même électrovalence, mais il peut y avoir substitutions multiples avec compensation électrostatique. La plus connue existe dans les feldspaths plagioclases, entre l'albite Na[AlSi3O8] et l'anorthite Ca[Al2Si2O8], le remplacement de Na+ par Ca2+ étant compensé par celui de Si4+ par Al3+ (cf. feldspaths).
- En général, les possibilités des substitutions augmentent avec la température de formation. Ainsi, à haute température ( > 700 °C), le sodium et le potassium sont totalement interchangeables dans la structure des feldspaths.
Lorsque la température retombera, la solution solide deviendra instable, il y aura précipitation d'un autre composé : exsolution. Le feldspath sodipotassique de haute température donnera par exsolution des associations orientées d'albite et de feldspath potassique K[AlSi3O8], connues sous le nom de perthites.
La température d'exsolution donne ainsi une limite inférieure de température de formation de l'association étudiée (thermomètre géologique).
Dans le cas des solutions solides d'addition, les atomes substitués occupent des positions interstitielles dans le réseau de l'hôte. Ces solutions solides sont fréquentes dans les phyllosilicates et tectosilicates : minéraux des argiles et zéolites.
Les solutions solides de soustraction constituent le dernier type. Dans la pyrrhotine, Fe1-xS, les analyses montrent toujours un excès de soufre qui est dû à l'absence de certains atomes de fer dans le réseau, mais, pour assurer la nécessaire compensation électrostatique, certains atomes de fer du réseau deviennent trivalents.
Le problème des éléments en traces
La plupart des analyses de minéraux montrent la présence de nombreux éléments en traces (teneur inférieure à 1%). Depuis le développement de la géochimie, on considérait que ces éléments en traces résultaient le plus souvent de substitutions diadochiques.
Des études, permises surtout par l'emploi de la microsonde électronique, réalisant des analyses ponctuelles sur des échantillons homogènes, à l'échelle des quelques micromètres cubes de volume intéressé (cf. infra. Méthodes quantitatives), ont montré que les substitutions diadochiques sont rares en dehors des solutions solides classiques.
Ce sont plutôt les défauts, les microfissures, les inclusions qui régissent la présence des « éléments en traces », qui sont en réalité fréquemment des « éléments fissuraux », situés en dehors du réseau du minéral hôte. Il convient donc maintenant de préciser par des méthodes annexes la forme cristallochimique d'un élément avant de considérer qu'il est substitué diadochiquement.
Polymorphisme
Plusieurs espèces minérales sont polymorphes si elles ont même formule chimique, mais des structures différentes ; pour un élément on emploie le terme d'allotrope :
par exemple, diamant et graphite sont deux formes allotropiques du carbone ; aragonite et calcite sont deux espèces polymorphes du carbonate de calcium CaCO3.
Les différents polymorphes d'une même substance sont formés sous différentes conditions de température, de pression, d'environnement chimique ; donc la présence d'un polymorphe dans une roche renseigne sur les conditions de formation de cette roche. Si les conditions changent, il peut y avoir réarrangement du réseau et passage d'un polymorphe à un autre : transition. Le point de transition pour une pression donnée correspond à la température d'équilibre des deux phases.
Toutes les transitions sont réversibles, mais il peut y avoir des retards au changement d'état qui peuvent se chiffrer en millions d'années ou ne pas être décelables pour des températures ordinaires.
Les vitesses de transformation d'une espèce polymorphe en une autre sont très variables et dépendent du degré de reconstitution de la structure : si la transition correspond à une simple déformation du réseau, elle est pratiquement immédiate à la température du point de transition (transition du quartz (3 de haute température, hexagonal, en quartz a de basse température à 573 °C).
On peut considérer, par contre, des transitions comme pratiquement irréversibles, car demandant de fortes dépenses d'énergie pour détruire l'ancienne structure et en reconstruire une nouvelle (transition graphite = diamant).
Pseudomorphoses
Un minéral peut en remplacer un autre en en conservant la forme extérieure ; c'est une « pseudomorphose « : par exemple, un octaèdre de cuprite Cu2O peut se transformer plus ou moins complètement en malachite Cu2CO3(OH)2 ; la pyrite FeS2 peut se transformer en goethite FeO(OH).
Les pseudomorphose peuvent aussi résulter d'incrustation plus ou moins prononcée d'un minéral par un autre : quartz pseudo-morphosant la fluorine, calcification des sources incrustantes.
Enfin un minéral inclus dans une roche peut disparaître, laissant une cavité correspondant à son faciès cristallin, cavité qui peut être remplie par un autre minéral, cas fréquent de pseudomorphose de sel gemme en calcite.
Minéraux amorphes
La spécificité des minéraux amorphes est souvent mise en question ; en effet leur composition chimique est mal définie et ils ne sont que peu structurés. Les minéraux amorphes comprennent les verres, les gels et les minéraux métamictes.
Verres et gels
On connaît des verres de silice (90 à 99% de SiO2) appelés lechatelierites ; ils forment en particulier les fulgurites, tubes de longueur variable (quelques décimètres à quelques mètres), produits de fusion par la foudre du sable des plages ou des déserts ; ces verres existent aussi dans certains cratères d'impacts météoritiques.
Les gels proviennent de la solidification de solutions colloïdales. L'opale, gel de silice hydratée, est le plus commun ; on connaît des gels de sulfure (métastibnite : sulfure d'antimoine), d'oxydes (limonite, gummite), de phosphates (diadochite : phosphate de fer), de silicates (allophanes, chrysocolle).
Minéraux métamictes
Les minéraux métamictes sont optiquement isotropes et amorphes aux rayons X, d'éclat vitreux ou résineux ; sans clivage, ils ont une cassure conchoïdale. Ils sont toujours radioactifs, et c'est la radioactivité a de l'uranium ou du thorium qu'ils contiennent qui a provoqué la destruction de leur réseau cristallin.
Cette métamictisation est plus ou moins complète ; par chauffage, ils peuvent recristalliser en émettant de la chaleur. On citera : gadolinite Y2Be2Fe [OSiO4]2, thorite Th[SiO4], fergusonite YNbO4, euxénite (niobotantalotitanate de terres rares, avec uranium).
3. Minéralogie déterminative
II faut distinguer la description et la détermination d'une espèce minérale. Décrire revient à étudier toutes les propriétés mécaniques, physiques et chimiques, et à préciser les conditions de formation.
La minéralogie déterminative, par contre, peut être définie comme la science et l'art d'identifier un minéral à partir de certaines de ses propriétés. Le nombre des espèces minérales est relativement limité (plus de 4 200 environ), mais les minéraux sont très polymorphes, d'où une identification parfois difficile.
On distinguera une minéralogie déterminative immédiate, permettant de reconnaître à vue ou avec des essais simples un assez grand nombre d'espèces, d'une minéralogie déterminative de laboratoire, permettant seule une certitude, mais demandant l'emploi de méthodes physiques ou chimiques parfois complexes.
Minéralogie déterminative immédiate
La minéralogie déterminative immédiate est fondée sur l'observation d'un certain nombre de caractères externes, ceux relatifs à la forme étant groupés sous la dénomination de « faciès » d'un minéral.
Faciès cristallin
La forme cristalline est parfois suffisante pour reconnaître un minéral cristallisé macroscopiquement (quartz, calcite...), mais, dans la nature, les cristaux sont souvent déformés ; ce sont des cristaux imparfaits dont les faces peuvent être striées, hérissées de pointements ou s'être développées très différemment les unes des autres, ce qui peut faire penser à une symétrie fausse ; d'autres déformations sont d'origine mécanique (cristaux de sel gemme déformés lors du plissement des argiles).
Il existe pour la plupart des espèces un faciès cristallin qui est prédominant pour une espèce. On distingue les faciès suivants :
- Isométrique : les dimensions des cristaux sont sensiblement égales suivant les trois directions de l'espace (galène, blende, magnétite...).
- Tabulaire : cristaux développés suivant deux directions ; en fonction de l'épaisseur, on distingue les faciès aplatis (barytine), folliacés (wulfénite), micacés (mica, autunite), etc.
- Allongé : cristaux développés suivant une seule direction ; en fonction du rapport longueur/diamètre, on a des faciès prismatiques (quartz, stibine), columnaire (manganite. tourmaline), aciculaire (scolécite, millérite), fibreux ou capillaire (amiante).
Il existe de nombreux faciès intermédiaires : lamellaire, c'est-à-dire aplati et allongé (disthène, gypse) ; en tonnelet (corindon) ; lenticulaire (gypse), etc.
Striations
De nombreux cristaux montrent des faces plus ou moins profondément striées ; ces stries sont formées par des arêtes et des sillons qui résultent d'oscillations entre la croissance de deux formes différentes ou bien de macles de croissance polysynthéti-ques (cf. infra. Associations de cristaux, agrégats).
Faces vicinales
Les faces principales d'un cristal peuvent présenter dans certaines espèces (quartz, fluorine, topaze...) de légères dénivellations, entourées de véritables arêtes, dont l'orientation ne diffère que très peu de celle de la face principale.
Ces « faces vicinales », ayant souvent des indices particulièrement complexes, sont utiles pour reconnaître certaines mériédries et, en particulier, distinguer les cristaux droits et gauches des mériédries énantiomorphes (fig. 2).

fig. 2 - Faces vicinales du quartz droit et du quartz gauche.
Imperfections
Les phénomènes d'adsorption liés à la croissance peuvent être la cause de nombreuses imperfections : cristaux dendritiques (les métaux natifs cubiques, or, argent, cuivre, se présentent souvent en individus squelettiques formés de petits cristaux soudés les uns aux autres) ou vacuolaires ; inclusions fluides et solides.
Les inclusions fluides peuvent être gazeuses ou liquides. Largement répandues, elles permettent de déterminer la température de formation du cristal et la physicochimie du milieu de croissance.
La plupart des cristaux contiennent des inclusions solides. On peut distinguer des inclusions formées avant le cristal ayant servi de support de cristallisation, orientées indifféremment par rapport au cristal, des inclusions de croissance comme, par exemple, les « fantômes » des cristaux de quartz, les « croix noires » de la chiastolite, variété d'andalousite.
Des cristaux peuvent se former en englobant des grains de sable ; c'est le cas des rhomboèdres de « calcite de Fontainebleau ». Enfin, d'autres inclusions solides peuvent résulter de phénomènes d'exsolution ou d'épitaxie (croissance orientée d'un minéral sur un cristal d'une espèce différente).
Force de cristallisation
On distingue des minéraux automorphes, capables de développer leur forme cristalline en luttant contre ceux qui les entourent (les grenats, par exemple), et des minéraux xénomorphes, qui se développent dans les espaces restés libres (tectosilicates dans les roches).
Les cristaux sont dits inclus quand ils sont englobés dans la gangue qui les entoure en conservant toutes leurs faces, et libres quand ils sont développés dans des cavités, fissures et géodes (cavités plus ou moins sphériques, tapissées de cristaux).
Taille des cristaux
Une même espèce peut donner des cristaux de taille variable, mais en général les grands cristaux bien formés sont exceptionnels. On peut cependant signaler quelques « monstres « : microcline de 10 m de diamètre et pesant plusieurs centaines de tonnes (Kaatiala, Finlande) ; biotite de 5 m de diamètre (Evje, Norvège) ; quartz de 4 X
2 m (Oural) ; béryl de 16 t (Albany, États-Unis) ; spodumène de 16 m de longueur, pesant 90 t (Keystone, États-Unis).
Associations de cristaux, agrégats
Les cristaux peuvent se grouper d'une façon désordonnée, au hasard des conditions du milieu, ou bien former des groupes ordonnés, géométriques ; parmi ces derniers, on ne fera que citer simplement les groupements parallèles du quartz, les cristaux polysynthétiques, les rosettes (cf. hématite), pour insister plus longuement sur les macles.
Les macles
Les cristaux de nombreuses espèces s'associent parfois suivant des lois définies, en donnant des groupements connus sous le nom de macles. Les macles peuvent être simples (deux cristaux) ou multiples. Si plusieurs individus se répètent alternativement suivant le même plan de macle, on a une macle « polysynthétique », d'aspect souvent lamellaire.
Dans certaines macles, on passe d'un individu à l'autre par rotation (souvent de 180°) autour d'un axe dit de macle. Dans d'autres cas, les deux cristaux sont symétriques par rapport à un plan de macle, plan réticulaire commun aux deux cristaux ; ce plan de macle correspond souvent au plan d'accolement des deux cristaux.
Suivant leur mode de formation, on distingue différentes sortes de macles :
- Macles de croissance. Elles résultent de la croissance simultanée des cristaux géométriquement associés.
On distingue les macles par accolement, comme celle de l'orthose, de celles par pénétration, comme celle de la staurolite. Ces macles de croissance peuvent former des groupements cycliques d'aspect souvent pseudohexagonal, comme c'est le cas pour l'aragonite.
- Macles de transformation. Lors d'une transformation polymorphique, le cristal peut, sans changer de forme extérieure, se transformer en un agrégat de plusieurs cristaux d'orientation complémentaire.
- Macles mécaniques. Dans de nombreux minéraux, le choc ou la pression détermine une déformation du réseau en faisant prendre à une partie de l'édifice cristallin une nouvelle position qui, par rapport à l'orientation d'origine, correspond à une macle.
Les macles présentent fréquemment des angles rentrants et montrent souvent une pseudosymétrie de degré de symétrie supérieur à celui de l'espèce (l'aragonite, orthorhombique, en macles pseudohexagonales).
Le faciès très caractéristique des macles facilite la reconnaissance de certains minéraux : cassitérite, rutile, cérusite, aragonite, feldspaths...
Faciès des agrégats
Les agrégats dont les grains ont une dimension proche du millimètre sont dits grenus, et micacés s'ils sont formés par de fines lamelles. Si les grains sont trop fins pour être distingués à l'œil ou à la loupe, le minéral est dit massif (cryptocristallin si, à fort grossissement, il se montre formé de grains cristallins).
Les minéraux peuvent être stalactiformes, en masses mamelonnées, réniformes. botryoïdales (en grappes), dont la texture interne est souvent fibreuse, parfois fibroradiée. en agrégats coralloïdes.
Si l'agrégat est formé de nodules plus ou moins sphériques, il va du « pisolitique » à l'« oolitique », en fonction de la diminution de la taille des nodules ; enfin, le faciès peut être terreux, pulvérulent.
Clivage et cassure
Un minéral peut se briser irrégulièrement (cassure), ou bien, préférentiellement, suivant des plans liés à la structure (clivage).
Les clivages peuvent être plus ou moins faciles, plus ou moins parfaits : gypse et mica présentent des clivages faciles et parfaits. La direction d'un clivage est donnée par l'indice de la face à laquelle il est parallèle, par exemple : clivage cubique (100) de la galène, clivage octaédral (111) de la fluorine.
Le clivage, qui est une caractéristique de certaines espèces, est utile à leur reconnaissance ; par contre, certains échantillons d'une espèce non clivable peuvent présenter de faux clivages, appelés plans de fracture préférentielle, dus souvent à un début d'altération ou coïncidant avec des plans de macle.
La simple cassure peut être relativement caractéristique. On distingue des cassures conchoïdale (quartz), esquilleuse (amphibole), lamellaire (brucite).
Ténacité et dureté
La ténacité est la résistance offerte par un minéral à être cassé, broyé ou coupé ; un minéral peut être cassant (cérusite) ou tenace (staurotite) ; il peut être malléable (métaux natifs), parfois sectile s'il peut être coupé en copeaux ; il peut être flexible s'il peut être courbé (gypse), et élastique si, après avoir été courbé, il revient à sa forme initiale (mica).
La dureté est la résistance à la rayure (ou la résistance de la structure aux déformations mécaniques) ; c'est une caractéristique utilisée depuis l'Antiquité. En 1822, Friedrich Mohs (1773-1839) proposa une échelle des duretés relatives, encore très utilisée : chacun des minéraux raye le minéral plus bas que lui dans l'échelle et est rayé par celui situé plus haut (tabl. 4).
|
1 |
talc (toucher gras) |
|
|
2 |
gypse (rayé à l'ongle) |
|
|
3 |
calcite (coupée au couteau) |
|
|
4 5 |
fluorine apatite |
facilement rayées au couteau |
|
6 |
orthose |
|
|
7 |
quartz |
|
|
8 |
topaze |
rayent le verre |
|
9 |
corindon |
|
|
10 |
diamant |
|
tabl. 4 - Échelle de Mohs.
Des méthodes modernes permettent de préciser les duretés relatives (scléromètres), mais la dureté est une grandeur anisotrope, variant avec les directions cristallographiques, et il est difficile de s'en servir comme caractéristique absolue. Il existe, de plus, une dureté apparente de certains agrégats friables : l'ocre rouge se raie à l'ongle, mais est formé de fins grains d'hématite rayant le verre.
Il faut bien distinguer ténacité et dureté : ainsi le diamant, le plus dur des corps naturels connus, peu tenace, se casse facilement grâce à son excellent clivage octaédrique ; par contre, la fibrolite, variété de sillimanite, de dureté 6,5, est très tenace et ses blocs sont presque incassables au marteau.
Éclat, couleur, transparence On distingue l'éclat métallique (pyrite) de l'éclat submétallique (blende) ; parmi les éclats non métalliques, reconnaissables surtout sur cassure fraîche, on reconnaît l'éclat vitreux (quartz), adamantin (diamant), résineux (soufre), nacré (brucite), soyeux (asbeste), terreux (agrégats à grain fin).
Les couleurs fort variables des minéraux n'aident que rarement à leur reconnaissance. De nombreuses espèces présentent une gamme de couleurs très complète (fluorine, corindon, quartz, béryl, tourmaline...).
Parfois la couleur est une propriété fondamentale liée à la composition chimique ; les ions ou groupes d'ions produisant une couleur caractéristique sont les « chromophores « : Cu2+ est le chromophore des minéraux secondaires bleus et verts du cuivre ; (UO2)2+ le chromophore des minéraux secondaires d'uranium, généralement brillamment colorés en jaune ou en jaune-vert.
D'autres fois, la couleur sera fonction du type de liaison structurale (diamant et graphite), des défauts structuraux, souvent provoqués par des radiations, ou bien elle proviendra d'impuretés liées ou non au réseau : ainsi de nombreux silicates des roches acides sont colorés en rouge par de microscopiques inclusions d'hématite.
La couleur de certains cristaux varie avec leur position par rapport à la direction d'observation. En lumière polarisée non analysée (lumière « naturelle » des pétrographes), ces minéraux « polychroïques » se reconnaissent aisément à leur pléochroïsme : variation de la couleur avec l'orientation du cristal par rapport au plan de polarisation du microscope.
La couleur de la poussière, beaucoup moins dépendante de l'état de surface et des impuretés, est un caractère de reconnaissance parfois très valable : on peut ainsi distinguer facilement l'hématite à poussière rougeâtre de la « limonite » à poussière jaune d'ocre, le cinabre à poussière vermillon du réalgar à poussière jaune orangé.
Un minéral sera transparent si l'on peut reconnaître un objet à travers, translucide s'il laisse passer la lumière, opaque enfin dans le cas contraire.
Caractéristiques diverses
La densité, propriété spécifique, ne peut que très grossièrement être évaluée à la main (cf. infra, Méthodes quantitatives. Densité). Des minéraux solubles, comme certains halogénures, nitrates, sulfates, ont un goût caractéristique ; c'est là une réaction chimique qualitative qui permet, par exemple, de différencier immédiatement la halite (NaCl) de la sylvite (KC1).
Les associations de minéraux ou « paragenèses » sont beaucoup plus importantes comme critères de reconnaissance. Certaines paragenèses sont en effet caractéristiques, par exemple l'association blende-pyrite-galène ; d'autre part, certains minéraux sont liés aux conditions d'équilibre ayant provoqué la formation de roches définies : minéraux du granité, minéraux des pegmatites, des roches basiques, etc.
Cela permet d'éviter des confusions, par exemple entre le grenat pyrope des serpentines et le grenat almandin des micaschistes.
Minéralogie déterminative de laboratoire
Méthodes qualitatives
Fluorescence : certains minéraux sont fluorescents sous l'ultraviolet ; ce caractère est très utilisé en prospection, par exemple pour rechercher la scheelite, CaWO4, minéral difficile à reconnaître autrement.
Radioactivité : compteurs de Geiger et scintillomètres sont d'un usage très fréquent pour reconnaître la présence d'uranium ou de thorium dans un minéral.
Magnétisme : le nombre des espèces minérales attirables par l'aimant est faible (7 espèces) ; c'est un test immédiat pour la magnétite, la pyrrhotine.
Analyse chimique : les minéralogistes utilisaient naguère des méthodes qualitatives basées sur l'emploi d'un chalumeau ; ils pouvaient ainsi faire agir une flamme très chaude sur un petit fragment de minéral et effectuer diverses opérations d'oxydation, de réduction, de sublimation qui leur permettaient, avec une grande habitude, d'obtenir assez rapidement une analyse qualitative partielle.
Actuellement, on utilise la microchimie qualitative, qui permet d'opérer sur de petites quantités (mm3) de minéral très pur. Le fragment est attaqué sous loupe binoculaire ; on ajoute ensuite des microgouttes de réactifs donnant, suivant les éléments présents, des précipités cristallins de forme caractéristique ou des colorations spécifiques.
On y adjoint souvent des microméthodes chromatographiques.
Méthodes quantitatives
Quand l'examen direct des caractéristiques externes des minéraux (morphologie, clivage, couleur, dureté) et l'analyse qualitative ne permettent pas de déterminer un minéral, il faut utiliser des méthodes quantitatives : mesure des propriétés optiques, densité, diagramme de Debye-Scherrer, analyses thermiques.
Ces méthodes sont généralement suffisantes, mais peuvent être complétées par la mesure des angles des faces cristallines et la détermination des rapports paramétriques, l'analyse structurale (groupe spatial et paramètres de la maille), l'analyse chimique quantitative.
Densité
La densité d (densités par rapport à l'eau ou densité usuelle) est le quotient de la masse (ou poids) du corps par la masse (ou poids mesuré au même lieu) d'un égal volume d'eau à 4 °C et à une pression de 760 mm de mercure.
La principale méthode utilisée est celle de la balance hydrostatique : le minéral est suspendu au plateau d'une balance ; on fait l'équilibre, puis on immerge le corps dans un liquide de masse volumique connue ; pour rétablir l'équilibre, il faut ajouter un poids représentant le poids d'un volume de liquide égal au volume du minéral.
Il est ainsi possible d'opérer sur 20 à 50 mg de minéral trié, avec des erreurs de 1 à 2 %. Mais il est parfois impossible de disposer de 20 mg du minéral pur.
C'est pourquoi les minéralogistes ont développé des méthodes sans pesées : on immerge un fragment de minéral dans un liquide de même densité, obtenu par mélange de différentes liqueurs lourdes ; si le corps reste en suspension, sa densité est égale à celle du liquide. On utilise le bromoforme (cl = 2,89), l'iodure de méthylène (d = 3,32), la liqueur de Clerici (formiate et malonate de thallium ; d = 4,5).
Diagrammes de Debye-Scherrer
Utilisés couramment, les diagrammes de poudre, véritables « empreintes digitales » des espèces minérales, ne suffisent pas toujours à déterminer avec certitude une espèce ; il faut en effet songer aux solutions solides dont les termes extrêmes peuvent donner les mêmes diagrammes.
De plus, des espèces différentes peuvent fournir des diagrammes presque identiques. Enfin, des minéraux en grains très fins dans des roches ou des minerais ne peuvent être extraits en quantité suffisante pour réaliser ces diagrammes. Il faut alors utiliser la diffraction électronique et les méthodes optiques de détermination (cf. pétrographie).
Analyses thermiques
L'analyse thermique différentielle (A.T.D.) et Y analyse thermopondérale, aujourd'hui couramment employées aussi bien en minéralogie qu'en chimie et en métallurgie, consistent à étudier un échantillon chauffé avec une vitesse de montée en température définie.
Dans l'analyse thermique différentielle, on enregistre à chaque instant la différence de température entre l'échantillon et un corps de référence chauffé simultanément.
Les variations brusques de cette différence marquent des transformations exo- ou endothermiques, et se traduisent par des crochets sur la courbe enregistrée. La température à laquelle se produisent les transformations, leur intensité, leur sens et leur vitesse (position, surface, forme des crochets enregistrés) constituent les caractéristiques thermiques du minéral.
Les effets endothermiques résultent de la perte d'eau de cristallisation, d'hydroxyle, d'anhydride carbonique ou de la destruction de la structure ; les effets exothermiques signifient oxydation ou recristallisation. Les courbes obtenues sont caractéristiques pour un grand nombre de minéraux. Cette méthode est particulièrement utilisée pour étudier les minéraux des argiles.
L'analyse thermopondérale est une technique complémentaire qui permet de mesurer en fonction de la température et du temps les variations du poids d'un minéral au cours de la chauffe.
Microsonde électronique de Castaing
Cet appareillage (1957) a révolutionné les possibilités de la minéralogie, de même que celles de la métallurgie. Il permet d'analyser quantitativement une « prise » de un cent-millionième de milligramme avec une limite de détection proche du cent-milliardième de milligramme.
On peut ainsi déterminer des espèces en grains très fins, incluses dans des roches ou des minerais, étudier facilement les phénomènes de diffusion et les anomalies de répartition des éléments majeurs ou en traces.
4. Classification des espèces minérales
Une classification doit rassembler des composés ayant des caractères communs.
Dans le système actuellement employé, les espèces sont groupées en classes en fonction de la nature des radicaux anioniques présents : 1) éléments natifs ; 2) sulfures et sulfosels ; 3) halogénures ; 4) oxydes et hydroxydes ; 5) carbonates, nitrates, borates ; 6) sulfates, chromâtes, molybdates, tungstates ; 7) phosphates, arséniates, vanadates ; 8) silicates ; 9) sels d'acides organiques, hydrocarbures, résines.
Les classes sont divisées en sous-classes d'après des caractères chimiques ou structuraux (par exemple sous-classe des tectosilicates) ; on trouve ensuite des groupes comprenant des espèces en étroit rapport (groupe des feldspaths, groupe des pyroxènes...).
Ces groupes peuvent être divisés en sous-groupes ; ainsi le groupe des pyroxènes comprend un sous-groupe orthorhombique et un monoclinique. Les groupes et sous-groupes comprennent les espèces.
On distingue, de plus, des sous-espèces (généralement des divisions arbitraires dans les séries isomorphes : oligoclase, labradorite, etc. de la série des plagioclases, par exemple) et des variétés.
Les variétés peuvent avoir des propriétés morphologiques ou physiques distinctes - couleur (améthyste, rubis), faciès (calcédoine) - ou des compositions chimiques légèrement différentes (marmatite : blende ferrifère ; campylite : mimétite phosphatée).
À l'exception des noms anciens consacrés par l'usage - blende, galène, béryl... -, les noms de minéraux doivent se terminer en -ite. Les noms dérivés du latin et du grec sont souvent liés à un caractère de l'espèce : albite, disthène, barytine, calcite...
Mais les noms d'espèces dérivent surtout de noms de minéralogistes ou autres scientifiques : millérite (NiS), heulandite (zéolite), curite (uranate de plomb) ; du lieu d'origine : aragonite ; de la composition chimique : molybdénite.
Les variétés recevaient autrefois des noms spécifiques, d'où la complexité de la nomenclature minéralogique ; elles sont désignées par des adjectifs : ferrifère, sélénifère, arséniatée, calcique...
Aux plus de 4 200 espèces minérales reconnues s'ajoutent chaque année des espèces nouvelles.
Éléments natifs
Une soixantaine d'éléments et d'alliages existent à l'état natif (cf. éléments natifs) ; certains comme l'or, l'argent, le soufre, le carbone (graphite et diamant) ont une grande importance économique.
Les métaux natifs ont un fort éclat métallique, des densités élevées ; ils sont malléables et ne présentent pas de clivages. Ils sont souvent cristallisés en rameaux, en dendrites, en filaments.
Sulfures et sulfosels
Ils comprennent les sulfures, séléniures, tellurures, arséniures et antimoniures des métaux suivants : Ag, Cu, Zn, Pb, Fe, Co, Ni, Mo, W, Sn, Hg, As, Sb, Bi (cf. sulfures et sulfosels naturels). Les sulfosels, dont la minéralogie est très complexe, ont une formule générale du type AmBnXp : A = Cu, Ag, Pb, Sn ; B = As, Sb, Bi, Sn ; X = S.
Cette classe (près de 450 espèces) est très importante économiquement car elle renferme, surtout avec les sulfures, les espèces qui constituent les minerais de base pour tous les métaux autres que le fer, le manganèse, le zinc, le tungstène, l'or, l'argent et les métaux légers.
Sulfures et sulfosels ont un éclat généralement métallique : pyrite FeS2, galène PbS, stibine Sb2S3, molybdénite MoS2, argentite Ag2S, chalcocite Cu2S, pyrrhotite (ou pyrrhotine) Fe1-xS, bornite Cu5FeS4, chalcopyrite CuFeS2, mispickel (ou arsénopyrite) FeAsS, tétraèdrite Cu3SbS3, bournonite CuPbSbS3, etc. ;
rarement submétallique : blende (ou sphalérite) ZnS, covellite CuS, cinabre HgS, proustite Ag3AsS3, pyrargyrite Ag3SbS3, etc. ; exceptionnellement résineux : réalgar AsS, orpiment As2S3, etc. Ils ont souvent une forte densité. On les exploite généralement dans des gisements où ils ont été concentrés par des processus hydrothermaux ou sédimentaires.
Halogénures
La classe des halogénures comprend 160 espèces environ. Les chlorures et les fluorures sont de loin les plus abondants (cf. CHLORURES NATURELS et FLUORURES NATURELS) ; ils sont généralement vitreux, souvent peu colorés, de dureté et densité faibles.
Les fluorures, comme la fluorine (ou fluorite) CaF2 et la cryolite Na3AlF6, sont essentiellement d'origine profonde, alors que les chlorures, comme la halite NaCl et hsylvite KC1, se trouvent surtout dans des dépôts sédimentaires (évaporites). On rencontre aussi des chlorures, bromures et iodures de cuivre, d'argent (cérargyrite AgCl) dans les zones d'oxydation de certains gîtes.
Oxydes et hydroxydes
Parmi les quelque 250 espèces (cf. oxydes ET HYDROXYDES NATURELS),
On trouve surtout des composés ioniques. Cette classe comprend des oxydes multiples, qui, chimiquement, devraient être considérés comme des sels : spinelles (cf. spinelles), niobates, tantalates, certains titanates, uranates et antimoniates. Les oxydes de silicium (silice) sont, d'autre part, souvent rangés aux côtés des silicates (cf. infra, Silicates).
Les oxydes simples et multiples appartiennent généralement aux domaines du magmatisme, du métamorphisme et de la sédimentation marine :
hématite (ou oligiste) Fe2O3, corindon A12O3, ilménite (Fe, Mg) TiO3, pérovskite CaTiO3, cuprite Cu2O, zincite ZnO, spinelle MgAl2O4 magnétite Fe2+Fe3+2 O4, chromite FeCr2O4, columbite FeNb2O6, tantalite FeTa2O6, chrysobéryl BeAl2O4, bétafite U(Nb2Ti)O9, brookite, anatase et rutile TiO2, cassitérite SnO2, polianite et pyrolusite MnO2, quartz SiO2, uraninite UO2, thorianite ThO2, etc.
La majeure partie des hydroxydes se trouve dans les zones d'oxydation des gîtes métalliques et dans les roches altérées : gibbsite A1(OH)3, bœhmite et diaspore AIO(OH), brucite Mg(OH)2, goethite FeO(OH), manganite MnO(OH), psilomélane (Ba, Mn...)3 (OH)6.Mn8O16, opale SiO2.nH2O, etc. Oxydes et hydroxydes sont les principaux minerais de fer, chrome, manganèse, uranium, étain, aluminium, niobium, tantale.
Carbonates, nitrates, borates
Les carbonates (110 espèces environ ; cf. CARBONATES NATURELS) Ont souvent un Aspect pierreux. Ils sont incolores ou peu colorés, à l'exception de ceux du cuivre, du cobalt, du manganèse et de l'uranium.
Les carbonates hydratés [azurite Cu3 (OH)2 (CO3)2, malachite Cu2(OH)2 CO3, hydrozincite Zn5(OH)6(CO3)2] sont essentiellement liés au cycle exogène ; les carbonates anhydres, surtout sédimentaires [calcite et aragonite CaCO3, magnésite MgCO3, dolomite MgCa(CO3)2, rhodocrosite MnCO3, smithsonite ZnCO3, sidérite FeCO3, withe-rite BaCO3, strontianite SrCO3, cérusite PbCO3], peuvent cependant se rencontrer dans des gisements en liaison avec des roches éruptives.
Les nitrates sont rares (8 espèces) ; ils sont isostructuraux avec les carbonates et généralement solubles dans l'eau. Les plus importants sont le nitre ou salpêtre KNO3, en cristaux aciculaires, en croûtes, en efflorescences incolores à grises, et surtout la nitratine ou nitronatrite NaNO3, en masses grenues, incolores à blanches ; cette dernière est un minerai encore important.
Ces nitrates résultent de l'évaporation d'eaux circulant dans les sols de régions très arides. Les borates (une centaine d'espèces) ont une structure formée de groupes [BO3] ou de tétraèdres [BO4].
Les borates anhydres qui se rencontrent dans des roches pegmatitiques ou de métamorphisme de contact sont rares à la relative exception des minéraux de la série ludwigite (Mg, Fe2+)2Fe3+(O2 BO3) - vonsenite (Fe2+, Mg)Fe3+O2BO3) ; orthorhombique, cristallisée en lames ou prismes mal formés, en agrégats microcristallins à éclat vitreux ; couleur noire avec reflets verdâtres, avec magnétite et grenats dans des gîtes de contacts.
Les borates hydratés, d'éclat vitreux, incolores à blanc, ont une faible dureté et sont peu denses.
Les principaux activement exploités (1 million de tonnes d'équivalent B2O3, États-Unis, U.R.S.S., Turquie. Argentine) sont : la kernite Na2B4O7.4 H2O, en masses incolores, clivables, à structure fibreuse, Yulexite NaCaB5O9.8 H2O, en masses blanches noduleuses, formées de cristaux capillaires ; le borax Na2B4O7.10 H2O, en cristaux monocliniques, prismatiques, en masses compactes ; la colémanite Ca2B6O11.5 H2O, en cristaux monocliniques lenticulaires, riches en faces, incolores à blancs.
Les borates hydratés se trouvent dans des bassins d'évaporation d'eau ayant lixivié des roches volcaniques.
Sulfates, chromâtes, molybdates, tungstates
Les sulfates (près de 300 espèces ; cf. sulfates naturels) peuvent être anhydres, comme la barytine BaSO4, Yanhydrite CaSO4, Yanglésite PbSO4, la célestine (ou célestite) SrSO4, souvent d'origine hydrothermale ; plus fréquemment hydratés, comme le gypse CaSO4.2 H2O, la bro-chantite Cu(SO4).3 Cu(OH)2, Yepsomite MgSO4.7 H2O, Y alunite KA13(OH)6(SO4)2, la kàînite MgSO4.KC1.3 H2O, ce sont des minéraux secondaires trouvés dans des zones d'oxydation ou des dépôts d'évaporation.
Sans éclat métallique, ils ont faibles densité, dureté et biréfringence.
Les chromâtes et les molybdates (une vingtaine d'espèces) sont rares, à l'exception de la crocoïte PbCrO4, en prismes monocliniques, rouge-orangé, et surtout de la wulfénite PbMoO4, en cristaux quadratiques tabulaires ou lamellaires, dense (d = 6,9), très fragile, jaune-orangé-rouge à brun, translucide, à éclat résineux, poussière incolore.
La wulfénite se rencontre surtout dans les zones subtropicales, avec cérusite, pyromorphite, comme minéral secondaire de la zone d'oxydation de gîtes plombifères.
Les tungstates (une quinzaine d'espèces) comprennent deux espèces très importantes, seuls minerais de tungstène :
la wolframite (Fe,Mn)WO4 et la scheelite CaWO4. La wolframite est un terme moyen d'une série isomorphe dont les termes extrêmes sont peu fréquents : ferbérite FeWO4 - hùbnérite MnWO4.
Elle se présente en cristaux monocliniques prismatiques, dépassant parfois le décimètre, souvent aplatis, généralement en masses grossièrement lamellaires, montrant un bon clivage parallèle à l'allongement ; sa densité est de 7,3, sa dureté de 4,5, sa couleur gris-noir à noir de fer (les termes riches en manganèse sont brun plus ou moins clair), son éclat métallique, sa poussière brun rougeâtre.
Elle se trouve dans des gîtes pneumatolytiques (pegmatites et greisen), avec quartz, tourmaline, lépidolite, mispickel, cassitérite, ou dans des filons hydrothermaux de haute température.
La scheelite se rencontre en octaèdres quadratiques parfois centimétriques ; elle est souvent massive à grenue ; sa densité est égale à 6,1 et sa dureté égale à 4,5 ; elle est blanchâtre-jaunâtre, translucide ; son éclat est gras, sa poussière incolore ; elle présente une vive fluorescence en blanc bleuâtre aux rayons ultraviolets.
On la trouve dans les gîtes de métamorphisme de contact, entre granité et roches calcaires (skarns et tactites), et parfois dans des filons hydrothermaux, avec la wolframite.
Phosphates, arséniates, vanadates
Les unités structurales de ces minéraux (près de 450 espèces) sont des tétraèdres [XO4]3: X = P, As, V.
La plus grande partie du phosphore se trouve dans la nature sous forme d'apatite Ca5(PO4)3(F, Cl, OH). L'arsenic des arséniosulfures et des arséniures donne des arséniates dans les zones d'oxydation, où l'on rencontre aussi les vanadates résultant de concentrations secondaires à partir du vanadium dispersé dans les roches et les minerais.
Dans cette classe, les minéraux primaires sont presque uniquement des phosphates non hydratés, déposés lors des phases terminales des processus magmatiques ; la plupart des autres espèces (arséniates, vanadates, phosphates hydratés), souvent rares, sont en effet d'origine secondaire et se trouvent dans les zones oxydées.
Espèces primaires
Les espèces primaires les plus importantes sont :
- Yamblygonite (Li, Na)AlPO4(F, OH), en masses clivables ou compactes ressemblant aux feldspaths, trouvée dans les pegmatites riches en lithium ;
- la triplite (Mn, Fe)2PO4(F, OH), en masses clivables, brun-noir à rougeâtre, d'éclat résineux, trouvée dans les pegmatites granitiques et donnant par altération de très nombreux phosphates hydratés ;
- la lithiophyllite Li(Mn, Fe)PO4, rare en cristaux orthorhombiques, surtout en masses clivables gris verdâtre, d'éclat vitreux, trouvée dans les pegmatites granitiques ;
- la monazite (Ce,La,Y,Th)PO4, monoclinique, en cristaux tabulaires, souvent en grains dans les alluvions, densité 5,2, clivable, brun caramel à brun-rouge, d'éclat résineux, radioactive, élément accessoire fréquent des granités, syénites et pegmatites alcalines, et important minerai de thorium et de terres rares.
Il faut citer également : le xénotime YPO4, la lazulite (Fe, Mg)Al2(PO4)2(OH)2.
Espèces secondaires
On recense de nombreuses espèces d'origine secondaire, dont les plus connues sont :
- Yolivénite Cu2AsO4(OH), fréquente en petits cristaux orthorhombiques allongés, vert olive à vert noirâtre, dans la zone d'oxydation des gîtes de cuivre à sulfoarséniures ;
- la scorodite FeAsO4.2 H2O, en octaèdres orthorhombiques, surtout en croûtes, en masses grenues à terreuses, vert pâle à vert foncé, d'éclat vitreux, minéral fréquent dans la zone d'oxydation des gîtes contenant du mispickel ;
- Yérythrite
(ou érythrine) Co3(AsO4)2. 8 H2O, en cristaux
lamellaires à aciculaires, en croûtes fibreuses à terreuses, possédant un
clivage parfait, rosé à rose-carmin, formée par oxydation des arséniures et
sulfoarséniures de cobalt ;
- la vivianite Fe3(PO4)2.8 H2O, fréquemment en prismes allongés, monocliniques et pouvant atteindre un mètre et plus, ou en agrégats fibreux, croûtes cristallines, de densité 2,6, sectile, au clivage parfait, vert-bleu à bleu-noir, translucide, donnant une poussière incolore, bleuissant à l'air, minéral fréquent dans les chapeaux de fer et dans les zones d'altération des pegmatites, dans les argiles, les sédiments à glauconie, les lignites ;
- la phannacolite CaHAsO4.2 H2O, en petits cristaux aciculaires, en croûtes fibreuses, blanche, produit d'oxydation récente des minéraux arsénifères ;
- la descloizite (Zn, Cu)PbVO4(OH), en pyramides orthorhombiques, en croûtes à pointements cristallins, relativement dense (d = 6.1), brun-rouge à noir, d'éclat gras, présente dans la zone d'oxydation des gîtes plombozincifères ;
- lapyromorphite Pb5Cl(PO4)3, la mimétite Pb,Cl(AsO4)3, la vanadinite Pb5Cl(VO4)3, qui appartiennent au groupe de l'apatite et entre lesquelles il existe des séries isomorphes plus ou moins complètes, fréquentes -surtout la pyromorphite - dans la zone d'oxydation des gîtes plombifères, souvent cristallisées en prismes hexagonaux, formant des masses grenues à aciculaires, de densité 7.2, de dureté 3,5, de couleur brun à vert (surtout pyromorphite), jaune (surtout mimétite), orange à rouge (surtout vanadinite), à éclat résineux à gras et poussière jaunâtre ;
la waxelïite A13(PO4)2(OH)3.5 H2O, en agrégats globulaires, croûtes, stalactites à structure fibroradiée, jaune-vert à vert, dans les fissures de roches alumineuses peu métamorphiques ainsi que dans des limonites phosphatées ;
la turquoise CuAl6(PO4)4(OH)8.4 H2O, en masses finement grenues à compactes, parfois conditionnées, de densité 2,7, de dureté 5,5, à cassure conchoïdale, bleu ciel ou bleu turquoise à vert, opaque, à poussière blanche, peu fréquente, minéral d'altération superficielle en climat aride dans les roches alumineuses d'origine éruptive ou sédimentaire ;
- Yautunite Ca(UO2)2(PO4)2.10 H2O, en cristaux quadratiques tabulaires à micacés, formant des agrégats écailleux à lamellaires, possédant un excellent clivage, jaune brillant à jaune verdâtre, fluorescente, minéral secondaire de la zone d'oxydation des gîtes uranifères, important minerai d'uranium ;
- la carnotite K2(UO2)2(VO4)2.3 H20, en cristaux orthorhombiques tabulaires formant des agrégats écailleux, des incrustations ou des masses pulvérulentes, de couleur jaune citron, minerai d'uranium ;
- la torbemite (ou chalcolite) Cu(UO2)2 (PO4)2.8-12 H2O, de même faciès que l'autunite, mais vert émeraude à vert noirâtre, non fluorescente et moins fréquente ;
- la francevillite (Ba,Pb)(UO2)2(VO4)2. 5 H2O, en cristaux lamellaires formant des groupes flabelliformes, des croûtes cristallines, jaune orange à jaune citron, clivable, non fluorescente, important minerai d'uranium.
Silicates
Les silicates, près de 900 espèces (cf. silicates), constituent, avec la silice (cf. silice), environ 95 % en poids de la totalité des minéraux, d'où leur extrême importance géologique (cf. amphiboles et PYROXÈNES, ARGILES ET MINERAUX ARGILEUX, CHLORITES, FELDSPATHOÏDES, FELDS-PATHS, GRENATS, MICAS, PERIDOTS).
Ils se reconnaissent à un éclat généralement vitreux, une forte dureté, une densité moyenne à faible (2,6 à 3,3), une poussière incolore à grise, même pour les silicates très colorés.
Mineraux lourds
Sels d'acides organiques, hydrocarbures, résines
Dans la plupart des cas, les nombreuses « espèces » décrites dans cette classe se révèlent être des mélanges mal définis de composés organiques ; c'est le cas en particulier des charbons, des ozocérites (paraffines) et de l'ambre (cf. ambre).
Comme espèces assez fréquentes, on peut citer : la mellite Al2(Ci2O12).18 H2O, en octaèdres quadratiques, jaune miel, trouvée dans certaines lignites ; la whewel-lite Ca(C2O4).H2O, en cristaux monocliniques, incolores, très fragiles, dans des filons de houille et rarement dans des gîtes hydrothermaux.
CLAUDE GUILLEMIN
Bibliographie
G. AlIBERT, C. GUTLLEMIN & R. PlERROT, Précis ck minéralogie, Masson, Paris, 1978 / A. Baronnet, Minéralogie, Dunod, Paris, 1988 / L. Berry & B. Mason, Mineralogy, Freeman, New York, 2e éd. 1983 / J. R. Clark & G. E. Brown, Chemistry and Physies of Minerais, Polycrystal Book Service, Dayton (Ohio), 1979 / W. A. Deer, R. A. Howm & J. Zussman, Rock-Forming Minerais, 5 vol., Halsted Press, New York, 2' éd. 1982-1986 / R. Evans, Crystal Chemistry, Univ. Press, Cambridge (Mass.), 1964 / M. Fleischer, Glossary of Minerai Speeies, Mineralogical Record, Tucson, 1983 / K. Frye, Minerai Science : an Introduction Survev, Macmillan Publ., New York, 1993 ; The Encyclopedia of Mineralogy, Stroudsburg (Pa.), 1981
W. Fyfe, Geoche-mistry of Solids, Me Graw-Hill, New York, 1964 / R. Gay, Cours de cristallographie, Gauthier-Villars, Paris, 1961 / D. T. Griffen, Silicate Ciystal Chemistry, Oxford Univ. Press, New York, 1992 / C. Guillemin & J. Mantienne, En visitant les grandes collections minéralogiques mondiales, B.R.G.M., 1988 / R. Hochleitner, Minéraux et cristaux, Nathan, Paris, 1987 / A. Lacroix, Minéralogie de la France et de ses anciens territoires d'outre-mer. 6 vol., Librairie du Muséum, rééd. Paris, 1979 / P. Laf-fitte, Introduction à l'étude des roches métamorphiques et des gîtes métallifères, Masson, 1957 / A. de Lapparent, Cours de minéralogie, ibid., 1899 / B. Masson, Principles ofGeochemistrv, Wiley & Sons, 1982 / P. Picot & Z. Johan, Atlas of are minerais.
Bureau de recherches géologiques et minières, Orléans, 1982 / R. Pierrot, Chemical and Determi-native Tables of Mineralogy, Masson, New York-Paris, 1979 / W. L. Roberts, Encyclopedia of Minerais, Van Nostrand, New York, 2e éd. 1989 / H. J. Schubnel, Larousse des minéraux, Larousse, Paris, 1981 / J. Sinkankas, Mineralogy for Amateurs, Van Nostrand, 1964 / G. Strtjbel & S. H. Zimmer, Lexikon der Minéralogie, Ferdinand Enke Verlag, Stuttgart, 1982 / H. Strunz, Mineralogischc Tabel-len, Geest & Portig, Leipzig, 1983 / J. Wyart, « Le Silicium dans la nature », in Traité de chimie miné-raie, Masson, Paris, 1965.
Minéraux lourds
On définit le groupe des minéraux lourds par leur densité supérieure à 2,87. Ce sont le plus souvent des silicates complexes inclus dans les sédiments et provenant directement ou indirectement de l'érosion de roches endogènes.
Les minéraux lourds étant relativement rares, il est nécessaire de les extraire du sédiment pour qu'ils puissent être observés soit par transmission au microscope polarisant, soit par réflexion au microscope métallographique, selon qu'ils sont transparents ou opaques. L'extraction des minéraux lourds du reste du sédiment se fait par des liqueurs denses, dont la plus couramment employée est le bromoforme (d = 2,87) : les minéraux légers, tels que le quartz, les feldspaths, les micas, y flottent tandis que les minéraux lourds coulent.
Diverses méthodes permettent de séparer des types de minéraux lourds entre eux. Ainsi des liqueurs de densité 3,3 permettront-ils de séparer les minéraux de densité supérieure à 3,3 et les autres.
On utilise aussi les propriétés magnétiques de certains minéraux. Un simple aimant recueille les minéraux les plus magnétiques, et le séparateur électromagnétique de type Frantz classe les minéraux selon des niveaux de magnétisme croissants.
En raison de leur grande résistance à l'abrasion, la tourmaline, le zircon et le rutile sont les minéraux lourds les plus fréquents dans les roches sédimentaires détritiques (minéraux ubiquistes). Les autres minéraux, plus fragiles, proviennent généralement de l'érosion des roches métamorphiques.
Si, à l'origine, les études de minéraux lourds se limitaient, avec plus ou moins de succès, à la détermination de l'origine des sédiments, il est apparu que l'on pouvait en tirer davantage d'informations dans d'autres domaines de la géologie grâce aux progrès de la méthode.
À partir des associations de minéraux lourds présents dans un sédiment, on peut restituer la direction et le sens des paléocourants qui alimentaient un bassin sédimentaire. En effet, en raison de phénomènes de dispersion au cours du transport, plus on s'éloigne de la source d'un minéral lourd, moins il est abondant.
C'est ainsi que, observant une diminution du pourcentage de grenat lorsqu'on s'éloignait du Massif armoricain, on a pu montrer l'origine armoricaine d'une partie des dépôts miocènes de la vallée de la Loire : les « faluns ». On peut aussi déceler l'existence de roches endogènes qui, après avoir affleuré et avoir subi l'érosion, ont été enfouies sous des sédiments protecteurs récents.
Enfin, la présence de un ou plusieurs minéraux lourds, dits « satellites ». dans les alluvions d'un cours d'eau peut être caractéristique d'un minerai donné. Ainsi, la prospection minière à la bâtée permet, par les études des alluvions de rivières actuelles, de remonter jusqu'à des gisements productifs.
JOCELYNE VUILLEUMIER
MINETTE DE LORRAINE
La minette est le nom local désignant le minerai de fer oolithique qui affleure, en couches stratifiées, dans la région limite entre la Lorraine et le Luxembourg, selon une large bande à l'ouest de la Moselle.
La formation ferrifère est interstratifiée dans le Jurassique. Elle date du Toarcien supérieur.
Cette série jurassique comprend, de haut en bas : des calcaires à Harpoceras humphresianum ; des calcaires à H. sowerbyi ; des marnes à H. murchisonae ; la formation ferrifère, d'une puissance totale de vingt à cinquante mètres, qui comprend plusieurs couches de minette et dont la partie supérieure correspond encore à la partie inférieure des niveaux à H. murchisonae, tandis que la partie inférieure appartient déjà à la partie supérieure des niveaux à Trigonia navis ; des grès à Trigonia navis ; deux niveaux argileux à H. striatulum ; des marnes micacées du Lias.
La formation ferrifère comprend de sept à huit couches, bien individualisées, dont la puissance manque de régularité et de continuité ; localement, les passages latéraux de la minette aux couches stériles ne sont pas rares.
Vers 1930, la puissance limite d'exploitation était de 1 mètre pour les couches calcaires et riches, et de 1,75 m pour les minerais siliceux et pauvres.
À l'analyse, la composition moyenne de la minette est, en pourcentage, la suivante : Fe, de 27 à 40 % ; SiO2, de 4 à 22 % ; A12O3, de 1 à 9 % ; CaO, de 3 à 19 % ; P2O5, de 0,5 à 1,7 %.
La faible teneur en fer est caractéristique de cette minéralisation, ainsi qu'un fort pourcentage de phosphore. Les teneurs en fer et en calcium varient en sens inverse. Le minerai est terreux.
Les couches de minerai sont rarement homogènes sur toute leur hauteur et se composent d'une série de bancs de qualités différentes, avec ou sans transition de l'un à l'autre.
Le minerai lui-même contient des veinules irrégulières d'argile ou de calcaire ferrugineux. Par endroits, il est très riche en fossiles (bois fossiles et grosses coquilles épigénisées en calcite), agglomérés souvent en lumachelles ; ce qui donne à la formation un caractère littoral très net. La proportion du phosphore est en étroite relation avec l'abondance des restes organiques.
La structure du minerai est oolithique. Chaque oolithe est formée de couches concentriques de limonite plus ou moins argileuse autour d'un ou de deux centres, qui peuvent être un résidu de chlorite, ou de sidérite, très rarement un reste organique (échinodermes, articles de crinoïdes, petits gastéropodes). La surface en est lisse, brillante et de couleur brune à jaune-rouge.
L'analyse des oolithes isolés donne 53 % de Fe. Le ciment qui les réunit est un mélange de calcaire, d'argile, de silicates de fer, voire de sidérite ; il devient siliceux par une plus grande prédominance des grains de quartz mélangés ; tantôt blanc, tantôt gris noirâtre, tantôt brun, tantôt vert, il est peroxyde et devient jaunâtre aux affleurements. La teneur en fer du ciment ne dépasse pas 35 %.
Au point de vue minéralogique, la limonite est le plus abondant des minéraux, les autres étant représentés par la sidérite, la chlorite (chamosite) et l'hématite.
La métallurgie des minerais riches en phosphore posait, au siècle dernier, des problèmes délicats : on sait, en effet, que la présence de phosphore dans la fonte rend celle-ci cassante.
En 1877, Thomas Sidney Gilchrist et son cousin Percy Gilchrist découvrirent une technique permettant l'affinage de telles fontes : c'est le procédé Bessemer basique, suivant lequel le phosphore est éliminé en présence d'un excès de chaux (qui provient, ici, du revêtement intérieur du convertisseur).
GUY TAMAIN

Recherche personnalisée
![]()